TL;DR (for B2B buyers)
- First classify the product: a medical / therapeutic LED device and a general beauty electrical appliance do not need the same evidence package.
- Do not stop at “FDA registered.” Ask for the FDA 510(k) number, covered models, предполагаемое использование, product codes, and clearance documentation.
- For regulated-market products, verify market records and system evidence separately: FDA 510(k), Health Canada MDL, TGA ARTG, CE/MDR route, Iso 13485, MDSAP, UL / МЭК 60601, МЭК 62471, and optical reports.
- For non-medical beauty electrical devices, CE / UKCA, FCC, ПОГОДА / Глобальный Марк, RCM, Rohs, EMC, adapter certificates, маркировка, and manual files may be more relevant than medical-device authorization.
- Before signing, match the document pack to the exact model, accessory set, целевой рынок, private-label changes, and claims your brand will actually use.
Who this guide is for: B2B brand owners, private-label buyers, дистрибьюторы, importers, clinic-channel buyers, and sourcing teams comparing LED face masks, red light panels, устройства для роста волос, pet therapy products, or other light-therapy devices before choosing an OEM / ODM manufacturer.
Who it is not for: end consumers looking for treatment advice or a home-use routine. This is a sourcing and compliance-verification guide, not medical advice.
If you are sourcing an LED therapy device for your brand, you have probably already opened several manufacturer pages. Many of them say similar things: OEM / ODM, FDA, CE, частная марка, fast delivery, factory direct.
The question is not whether a supplier can print your logo on a mask. The real question is whether that supplier can support your brand when the first product becomes a product line.
This guide focuses on the evidence layer behind an LED therapy device OEM decision: how to verify FDA 510(k), how to read model scope, how to check Health Canada MDL, Iso 13485, MDSAP, UL / МЭК 60601, CE / UKCA, FCC, ПОГОДА / Глобальный Марк, and TGA records, and what batch-level test files buyers should request before placing an order.
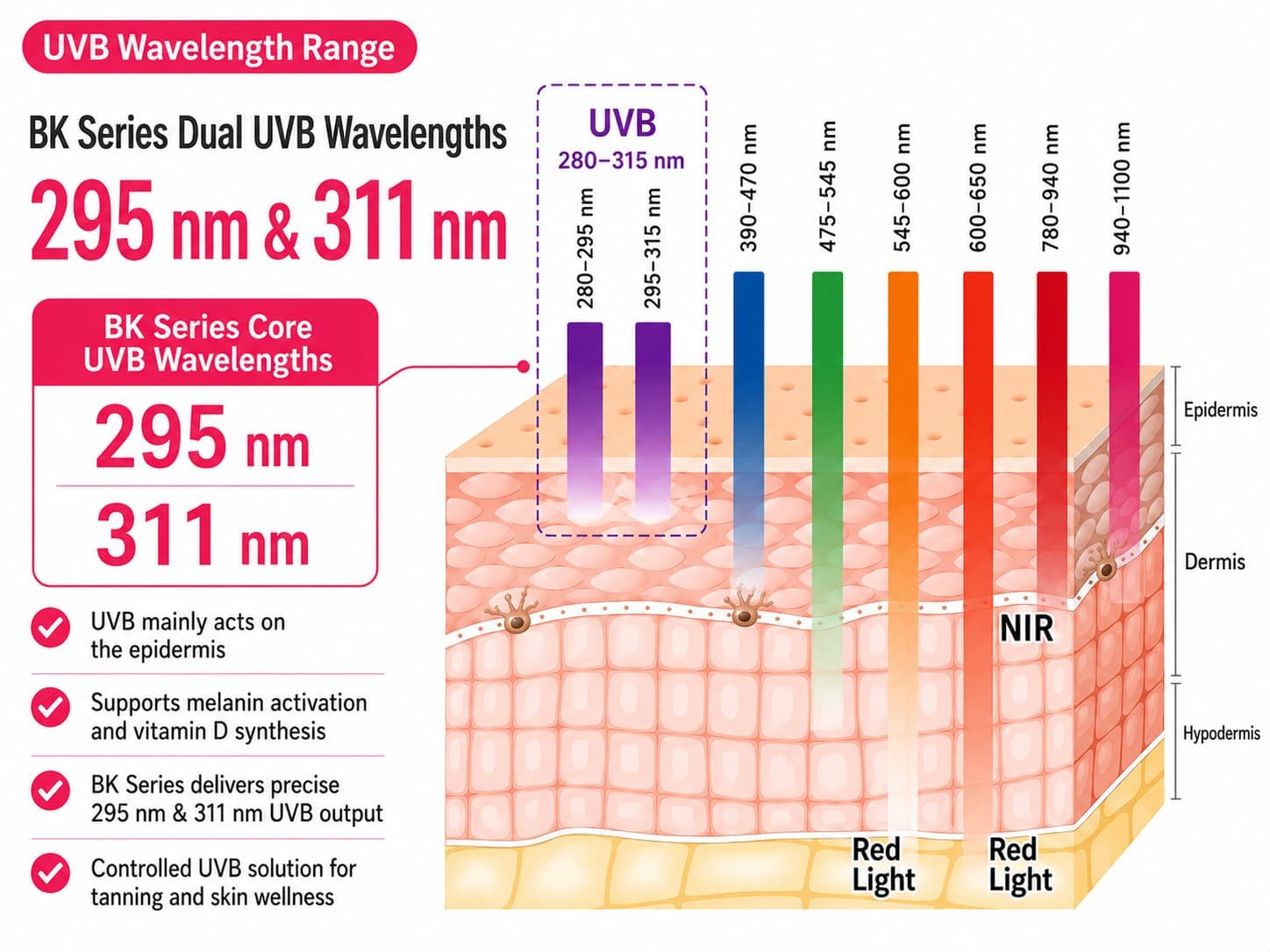
Before comparing certificates, define the product level. A red light beauty device positioned as a medical or therapeutic device may need market authorization, medical-device quality-system evidence, medical electrical safety review, and careful claim control. A general beauty electrical appliance or consumer wellness device may mainly need market conformity, EMC, электробезопасность, Rohs, adapter, маркировка, and product-safety documentation. The wrong shortcut is to treat every badge as the same kind of proof.
Use this decision map before requesting documents:
| First Question | If the answer is yes | Evidence Direction |
|---|---|---|
| Will the product be sold with медицинский, therapeutic, уход, облегчение боли, прыщи, wound-healing, or clinical-use claims? | Treat it as a regulated medical-device discussion until proven otherwise | Check market authorization, medical-device QMS, safety and performance evidence, маркировка, and claim review |
| Will the product be sold as красота, уход за кожей, Жестирочное, расслабление, or general red light exposure without regulated medical claims? | Treat it as a general beauty electrical / consumer wellness product discussion | Check CE / UKCA, FCC, EMC, Rohs, RCM / EESS, ПОГОДА / Глобальный Марк, adapter, электробезопасность, маркировка, and manual files |
| Will the same hardware be sold in several markets or under private label? | Do not rely on one badge list | Check model scope, accessory scope, importer / responsible-party obligations, label changes, manual changes, and claim changes |
Company names are quoted as shown in each official database. Capitalization and punctuation may differ between FDA, БСИ, TGA, and Health Canada records.
FDA-Cleared vs FDA-Registered: What Is the Difference, and Why Does It Matter?
FDA registration or listing means a company or device is recorded with the FDA; FDA 510(k) clearance means the FDA found a specific device substantially equivalent to a legally marketed predicate device for its stated indications. 510(k) clearance is not FDA approval.
This distinction matters because “FDA” is often used loosely in sourcing conversations. A factory may be FDA-registered, a device may be listed, a product may be 510(k)-очищен, or a product may be FDA-approved. These are different regulatory states, and they should not be mixed in one claim.
What “FDA registered” usually means
For medical devices sold in the United States, establishment registration and device listing are administrative requirements. They show that a company and listed device information are recorded with the FDA. They do not mean the FDA has cleared, approved, tested, or endorsed the product.
This is why a buyer should not ask only, “Do you have FDA?” A better question is:
What is the 510(k) number, and which exact device models does it cover?
Что FDA 510(k) clearance means
For a 510(k), the submitter provides a premarket notification showing that the device is substantially equivalent to a legally marketed predicate device. Depending on the device, documentation may include device specifications, performance testing, electrical safety and EMC testing, photobiological safety testing, biocompatibility information, маркировка, and predicate comparison.
The FDA may request additional information before issuing a decision. If the device is cleared, the database entry shows a 510(k) number such as “K250830.” This is the number buyers can verify.
One more point: timeline and cost should be discussed carefully. The FDA publishes annual MDUFA fees, but total project cost depends on testing, consulting, документация, engineering work, and response cycles. A serious 510(k) project can be expensive and time-consuming, but the exact number should not be treated as a universal rule.
What this means for your brand
If a supplier only says “FDA registered,” ask for the K number. If they give you an establishment registration number instead, that is not a 510(k). If they say the application is “in process,” that cannot be verified in the public 510(k) clearance database.
For a brand selling into regulated or platform-sensitive channels, this is not a paperwork detail. It affects product claims, listing review, distributor confidence, and your own compliance risk.
How Many Mask Models Does One 510(k) Cover?
А 510(k) clearance applies to a specific device or device family. Broader model scope can make multi-SKU planning easier, but any new model, маркировка, claim, or market entry still needs regulatory review.
Many first-time buyers ask whether a supplier “has 510(k).” Fewer ask what the clearance covers. That second question is often more useful.
What “FDA registered” usually means
For medical devices sold in the United States, establishment registration and device listing are administrative requirements. They show that a company and listed device information are recorded with the FDA. They do not mean the FDA has cleared, approved, tested, or endorsed the product.
This is why a buyer should not ask only, “Do you have FDA?” A better question is:
What is the 510(k) number, and which exact device models does it cover?
Что FDA 510(k) clearance means
For a 510(k), the submitter provides a premarket notification showing that the device is substantially equivalent to a legally marketed predicate device. Depending on the device, documentation may include device specifications, performance testing, electrical safety and EMC testing, photobiological safety testing, biocompatibility information, маркировка, and predicate comparison.
The FDA may request additional information before issuing a decision. If the device is cleared, the database entry shows a 510(k) number such as “K250830.” This is the number buyers can verify.
One more point: timeline and cost should be discussed carefully. The FDA publishes annual MDUFA fees, but total project cost depends on testing, consulting, документация, engineering work, and response cycles. A serious 510(k) project can be expensive and time-consuming, but the exact number should not be treated as a universal rule.
What this means for your brand
If a supplier only says “FDA registered,” ask for the K number. If they give you an establishment registration number instead, that is not a 510(k). If they say the application is “in process,” that cannot be verified in the public 510(k) clearance database.
For a brand selling into regulated or platform-sensitive channels, this is not a paperwork detail. It affects product claims, listing review, distributor confidence, and your own compliance risk.
How Many Mask Models Does One 510(k) Cover?
А 510(k) clearance applies to a specific device or device family. Broader model scope can make multi-SKU planning easier, but any new model, маркировка, claim, or market entry still needs regulatory review.
Many first-time buyers ask whether a supplier “has 510(k).” Fewer ask what the clearance covers. That second question is often more useful.
Why model scope matters
А 510(k) decision is not a factory-level certificate. It is tied to the submitted device, предполагаемое использование, technological characteristics, маркировка, and supporting documentation. If a future product changes in a way that affects safety, effectiveness, предполагаемое использование, or technological characteristics, further regulatory analysis may be needed.
So the practical buyer question is:
Is this a narrow clearance for one mask, or a broader device family that supports several related models?
Broader scope does not remove regulatory responsibility. Это делает, однако, give your product team a stronger starting point when planning a mask lineup.
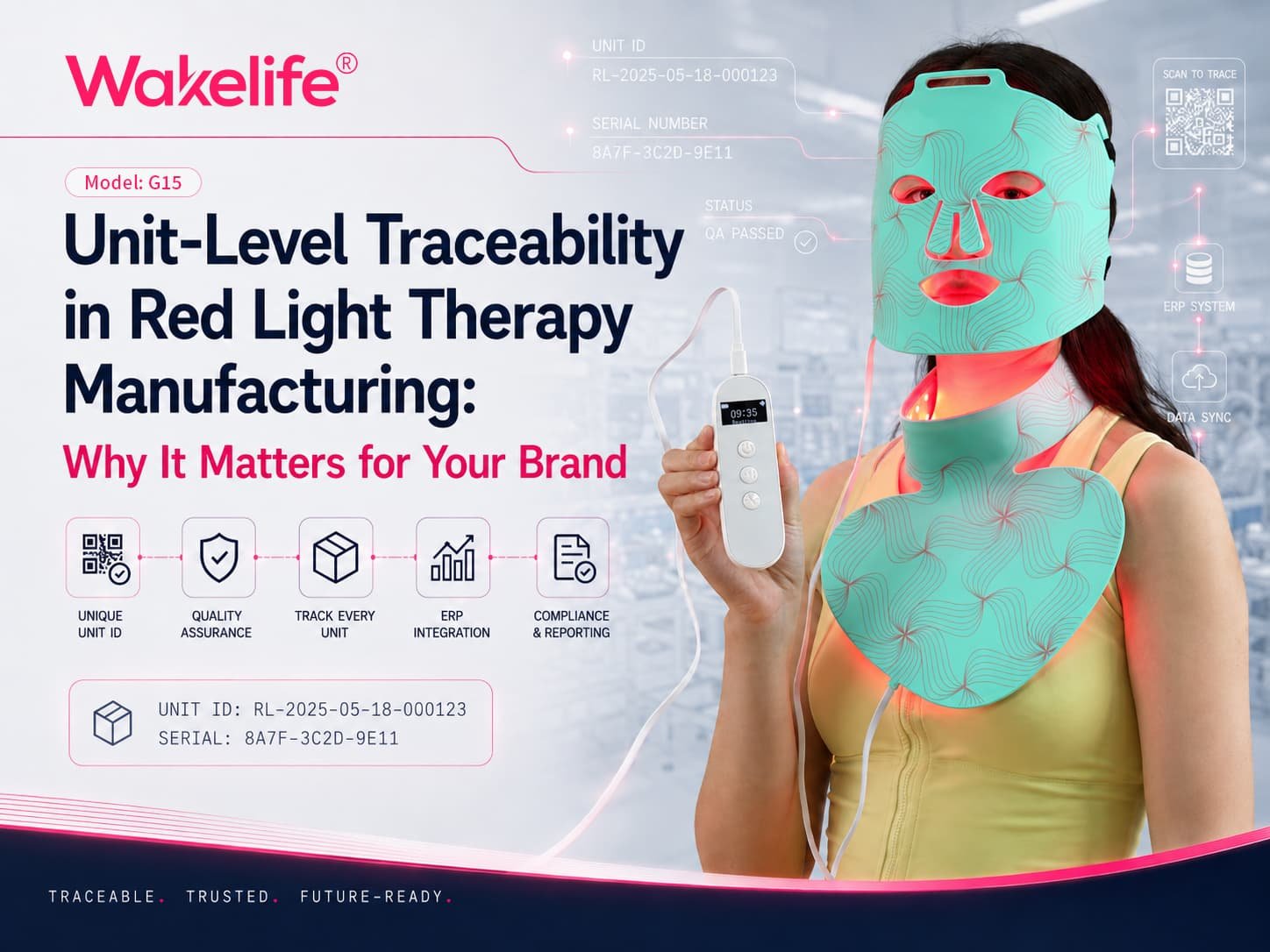
K250830: 13 mask models under one FDA clearance
On June 9, 2025, FDA 510(k) database shows K250830 для Шэньчжэньская компания Sungrow Led Technology Co., ООО., with the device name “LED Light Therapy Mask.” The database entry itself shows the applicant, device name, product codes (OHS and OLP), decision date, and a substantially-equivalent decision.
The clearance covers 13 модели, as documented in the clearance letter / 510(k) summary:
G15, G15p, Г15К, G11P, G11, G10, G13, G14, G17, ОБЩИЙ, ПРАНА, Chin2Chest, and BBL-FACEMASK.
Buyers can confirm the applicant, device name, product codes, and decision directly in the FDA 510(k) Premarket Notification database, and request the clearance letter to verify the full model list.
For private-label brands, this matters because a supplier with a broader cleared device family may support product-line planning better than a supplier with a single cleared SKU. The right interpretation is not “anything you launch is automatically cleared.” The right interpretation is: you have a clearer regulatory reference point for related models, and you can discuss the actual path with the manufacturer and regulatory consultant before launch.
Where Does Manufacturing Background Fit Into Compliance Evidence?
Factory size and category range are useful background, but they are not substitutes for public regulatory records, certificate numbers, and product-level test reports.
Manufacturing background still matters in supplier evaluation. A larger in-house facility and broader product capability may indicate stronger engineering resources, производственный контроль, and documentation discipline. But this type of information should be handled differently from third-party evidence.
Например, Wakelife describes its Shenzhen manufacturing facility as approximately 20,000 м² собственных производственных площадей on its manufacturing page. That is useful factory-background information. It should not be treated as the same evidence category as an FDA 510(k) record, a BSI certificate, or a TGA ARTG entry.
The clean way to read supplier evidence is to separate it into layers:
| Evidence Layer | Examples | How Much Weight to Give It |
|---|---|---|
| Public regulatory records | FDA 510(k), Health Canada MDL, TGA ARTG entries | Высокий: independently verifiable |
| Third-party QMS certificates | Iso 13485, MDSAP | Высокий: verify certificate number, scope, issuer, expiry |
| Regional conformity and EMC / electrical-safety evidence | CE / UKCA, FCC, ПОГОДА / Глобальный Марк, RCM, Rohs, adapter certificates | Important for market access and product safety, but do not treat as medical clearance |
| Product-level test reports | МЭК 62471, EMC, optical reports, biocompatibility where applicable | Высокий, but request the actual files |
| Factory background | Floor space, Производственные линии, category range | Useful context, but not a compliance claim |
| Sales materials | Brochures, website badges, catalog claims | Lowest unless backed by documents |
This separation keeps the article honest: manufacturing scale supports the supplier story, while public records and test files carry the compliance claim.
How Do You Verify a Manufacturer’s Certifications Without Relying on a Sales Deck?
Ask for identifiers that can be checked in public databases or official certificate directories. A logo on a website is not enough.
A serious supplier should be able to provide numbers, not just badges. Here are the checks a buyer can run before placing an OEM order.
1. FDA 510(k): verify the K number
Попросите 510(k) number, then search the FDA Premarket Notification database. Confirm:
- Applicant name
- Device name
- Decision date
- Decision outcome
- Product codes
- Model list, if shown in the entry or clearance documentation
For Sungrow/Wakelife’s LED Light Therapy Mask, the relevant entry is K250830.
2. Iso 13485: check certificate number, issuer, scope, and validity
Iso 13485 is a medical-device quality-management-system standard. It is not the same as product clearance. It tells you that the manufacturer’s quality system has been certified against the standard within a defined scope.
For Sungrow, the BSI client directory lists Iso 13485:2016 & EN ISO 13485, certificate number MD 800872, для Shenzhen Sungrow LED Technology Co.,LTD. The scope shown in the directory is: “The design, manufacture and distribution of LED therapy lights for skin treatment and pain relief and LED light therapy mask for the treatment of wrinkles and mild to moderate acne.”
When reviewing any supplier’s ISO 13485 сертификат, check:
- Certificate number
- Issuing body
- Company name and address
- Scope wording
- Effective date and expiry date
3. MDSAP: check whether the quality system has multi-market audit coverage
MDSAP stands for Medical Device Single Audit Program. It is a quality-system audit program recognized by participating regulatory authorities. It does not replace every country-specific market authorization, but it is meaningful evidence of a more mature quality system.
For Sungrow, the BSI client directory lists MDSAP 800873 для Shenzhen Sungrow LED Technology Co.,LTD. The directory entry shows original registration date 2024-05-14, last revision date 2026-04-08, and expiry date 2027-05-13.
4. TGA ARTG: check Australian medical-device listings
For Australia, buyers can verify ARTG entries through the TGA website. The following publicly verifiable face-mask records are used as worked examples in this guide:
| ARTG ID | Sponsor | Производитель | Название продукта / Тип | Сорт |
|---|---|---|---|---|
| 510468 ↗ | Apexmed Service Pty Ltd | Shenzhen Sungrow LED Technology Co Ltd | Cosmetic phototherapy system, для домашнего использования | Класс IIа |
| 525268 ↗ | Apexmed Service Pty Ltd | Shenzhen Sungrow LED Technology Co Ltd | Cosmetic phototherapy system, для домашнего использования | Класс IIа |
The point is not to decorate the page with certification logos. The point is to give your procurement, согласие, and distributor teams records they can verify independently. The public summaries do not, by themselves, establish that every Wakelife product, private-label model, claim, or configuration is covered. Buyers should confirm the applicable ARTG ID against sponsor/manufacturer scope documents for the exact product.
5. Health Canada MDL: check Canadian medical-device licensing
For Canada, buyers can verify active medical device licences through Health Canada’s Medical Devices Active Licence Listing (MDALL). The primary verification link should be the MDALL manufacturer page, not only one licence number.
The MDALL manufacturer page lists:
| Manufacturer Field | Verified Value |
|---|---|
| Производитель | SHENZHEN SUNGROW LED TECHNOLOGY CO., LTD ↗ |
| Адрес | 3f, 2# Заводской корпус, (Фаза I) Sci Carcass & Tech Ind Park, Guangming High-Tech Ind Park, Шэньчжэнь, 190, Китай, 518000 |
| Company ID | 187745 |
Buyers should then review each licence record separately. Например, the manufacturer page shows an LED mask record and an E-series therapy-light record.
| MDL Record | Public Details to Verify |
|---|---|
| LED LIGHT THERAPY MASK |
Licence No. 113279
| Device Family| Сорт 2
First Issue Date: 2025-05-09
Included Device Identifiers (MDALL):
BBL-МАСКА ДЛЯ ЛИЦАCHIN2CHESTG10G11G11PG13G14G15Г15КG15pG17ПРАНАОБЩИЙ
|
|
THE E-SERIES LED THERAPY LIGHT |
Licence No. 112124
| Device Family| Сорт 2
First Issue Date: 2024-11-04
Примечание: The MDALL device-identifier table should be reviewed directly on the live manufacturer page, as identifier lists are subject to updates over time.
|
These records should be read as Canada-specific market licence records. They should not be described as FDA clearance, TGA inclusion, or ISO/MDSAP quality-system certification. Also avoid writing that MDL 113279 covers panels; the mask record and the E-series therapy-light record should be checked as separate licence records under the same manufacturer page.
6. CE, UKCA, FCC, ПОГОДА / Глобальный Марк: check the non-medical conformity layer separately
Not every LED red light beauty device is marketed as a medical device. Some products are sold as general beauty electrical appliances or consumer wellness devices. For those products, the core documentation may be CE / UKCA, FCC, Rohs, EMC reports, electrical-safety reports, adapter certificates, RCM / EESS evidence, or SAA / Global-Mark certificates rather than FDA 510(k), Health Canada MDL, or TGA ARTG.
This layer is still important, but it proves a different thing. CE or UKCA documentation helps support applicable EU or UK product conformity. FCC evidence supports US electromagnetic compatibility or radiofrequency compliance. ПОГОДА / Global-Mark and RCM-related evidence may support Australia / New Zealand electrical-safety review, especially for powered devices, plugs, chargers, and adapters.
Buyers should check the declaration, отчет, or certificate by model number, стандартный, issuing body, report number, certificate number, tested configuration, and accessories covered. Do not treat CE, UKCA, FCC, ПОГОДА, Глобальный Марк, or RCM as proof of FDA clearance, clinical efficacy, or medical treatment claims.
For the EU specifically, buyers must distinguish general CE conformity from CE marking under Regulation (Евросоюз) 2017/745. If the device has an intended medical purpose—or may fall within an MDR Annex XVI product group—review the route, classification, Declaration of Conformity, and Notified Body scope in CE vs CE MDR for LED Beauty Devices.
7. UL 60601-1 и МЭК 60601: check medical electrical safety evidence
For powered LED therapy devices, buyers should also review electrical safety evidence. This matters most for products with adapters, chargers, rechargeable batteries, контроллеры, higher-power panels, or clinic-channel positioning.
МЭК 60601-1 belongs to the medical electrical equipment standards family and addresses general requirements for basic safety and essential performance. Whether it applies depends on the product’s intended use, нормативная классификация, целевой рынок, and any applicable particular standards; it is not a blanket requirement for every powered LED product. UL Solutions describes IEC 60601 testing and certification as a way to evaluate compliance with the safety and performance requirements for medical electrical equipment. In North America, certification or listing to an applicable standard by a recognized testing body, for example an NRTL under the OSHA NRTL program, can be meaningful safety evidence when required by the product, workplace, покупатель, or market pathway.
Unlike FDA 510(k), Iso 13485, MDSAP, TGA ARTG, or Health Canada MDL, electrical-safety certification does not always resolve to a single public database entry that buyers can search by company name. That is exactly why buyers should ask for the specific file or report identifiers and confirm them with the issuing body. Treat a UL or ETL logo on a page the same way you treat any other logo in this guide: unverified until you have the number behind it.
When reviewing UL / МЭК 60601 claims, ask for:
- Exact standard number and edition
- Test laboratory or certification body, such as UL, Intertek/ETL, TUV, or another recognized body
- Report number, certificate number, CB report, UL file number, or NRTL listing details
- Product model(с), adapter, charger, батарея, контроллер, and accessories covered
- Whether the file covers electrical safety, EMC, или оба
- Whether the tested configuration matches the commercial product you plan to order
Buyers should not treat UL or IEC 60601 evidence as FDA clearance, Health Canada MDL, TGA inclusion, or clinical efficacy evidence. It belongs to the electrical-safety layer, and it should be verified by file number, not by logo.
8. Batch-level optical records: ask for the data behind the product
Certifications do not replace production QC. For LED therapy devices, buyers should ask for recent batch-level records such as:
- Spectrophotometer wavelength verification
- Irradiance measurements with distance and test conditions
- МЭК 62471 photobiological safety reports
- Electrical safety and EMC reports where applicable
- Biocompatibility documentation for skin-contact materials where applicable
If a supplier can show current optical test records quickly, that is a good sign. If every answer becomes a brochure, keep asking.
How Should You Summarize the Evidence Before an OEM Decision?
Create an evidence table that separates public records, third-party certificates, product test reports, and factory-background information.
A useful evidence table keeps the conversation factual. Instead of asking whether a supplier is “certified,” list each claim beside the document or database record that supports it.
| Evidence Area | What to Ask | Красный Флаг |
|---|---|---|
| FDA status | What is the 510(k) number? Which models are covered? | «FDA registered» with no K number |
| Model scope | How many mask models are included in the cleared family or technical file? | One SKU with vague claims about future variants |
| Quality system | Iso 13485? MDSAP? Certificate number, issuer, scope, expiry? | Certificate logo with no verifiable number |
| Market records | TGA ARTG, Health Canada MDL, UKCA, CE documentation as relevant to your market | One generic certificate used for every market |
| General product compliance | FCC, ПОГОДА / Глобальный Марк, RCM / EESS, Rohs, EMC and adapter evidence where relevant | Electrical or EMC documents presented as medical authorization |
| Optical QC | Recent wavelength and irradiance reports with test conditions | Marketing specs but no batch data |
| Product test reports | МЭК 62471, optical data, EMC, material files where applicable | Brochure specs without reports |
| Factory background | Facility page, production capability, product categories | Factory-size claims with no context |
A restrained way to read certification breadth
For Wakelife/Sungrow, the strongest publicly verifiable records for this article are:
| Evidence Type | Public Identifier |
|---|---|
| FDA 510(k) | K250830 ↗ |
| Iso 13485 | BSI certificate MD 800872 ↗ |
| MDSAP | BSI certificate MDSAP 800873 ↗ |
| Health Canada MDL | MDALL Manufacturer Page — Company ID 187745 ↗ |
| TGA ARTG | 510468 ↗ | 525268 ↗ |
| Factory Background | Wakelife Manufacturing Page ↗ (Примерно 20,000 m² in-house manufacturing space) |
This does not mean Wakelife is automatically the right supplier for every brand. It means the evidence can be checked. The sourcing decision still depends on target market, claims, бюджет, график времени, Могил, потребности в настройке, and product roadmap.
What Evidence Should You Request Before You Sign?
Before paying a mold fee or confirming packaging, ask the supplier for a document pack:
- FDA 510(k) number and clearance letter, if the product is claimed as 510(k)-очищен
- Iso 13485 certificate with issuer, scope, and expiry
- MDSAP certificate, if relevant to your markets
- TGA ARTG, Health Canada MDL, UKCA, CE, FCC, ПОГОДА / Глобальный Марк, RCM / EESS, or other market documentation where relevant
- МЭК 62471 photobiological safety report
- Skin-contact material documentation and biocompatibility files where applicable
- Recent wavelength and irradiance test records
- Упаковка, маркировка, руководство, and claim-review support
- Product roadmap: маски, neck/chest devices, панели, рост волос, pet therapy, or other planned categories
This is also where supplier fit becomes clear. A good OEM partner will not only say “yes.” They will tell you which claims are safe, which documents are available, which claims need legal review, and which product ideas may require further testing or regulatory work.
If you are still defining the product itself, start with the LED Face Mask OEM Sourcing Guide first, then return to this certification guide when you are ready to verify documentation.
Request an OEM consultation: https://wakelifebeauty.com/contact/
Send your target market, intended product model, planned claims, expected order quantity, and next-category roadmap. We can help you compare the documentation you should request from any manufacturer, including suppliers that are not us.
Because the right manufacturer for your brand is not always the one with the loudest page. It is the one whose evidence can survive your distributor, platform, and compliance review.
Which Documents Matter Most for Each Target Market?
Start with the market where you will actually sell, then match documents to the product level, claim, model, and accessory set. A long certificate list is less useful than the right file for the right market.
Use this as a first-pass map before sending an RFQ. It is not a substitute for legal or regulatory review, but it helps buyers ask more precise questions.
| Целевой рынок | If Positioned as Medical / Therapeutic | If Positioned as General Beauty / Оздоровление | What to Ask the Supplier First |
|---|---|---|---|
| Соединенные Штаты | FDA 510(k) где это применимо, FDA registration / listing context, labeling and intended-use review, electrical safety and EMC evidence where relevant. | FCC / EMC evidence, electrical safety files, adapter / charger documentation, Rohs / Prop 65 review where relevant, claim substantiation. | «Is this exact model covered by a 510(k), or only FDA registered / listed? If non-medical, what FCC / EMC and electrical-safety files cover it?» |
| Евросоюз | CE marking under MDR where the device is medical or follows an applicable MDR route; classification, technical documentation, DoC, Notified Body status where required. | Общий CE route: DoC, EMC, LVD where relevant, Rohs, RED for wireless functions, technical file, маркировка. | «Is this general CE documentation or CE marking under MDR? Which EU legislation and standards are listed on the DoC?» |
| Великобритания | UK medical-device route / UKCA or accepted CE route depending on current rules and product status; UK responsible-party review where applicable. | UKCA / accepted CE документация, EMC, электробезопасность, Rohs, radio evidence where applicable, UK labeling review. | «Which UK route is being used, and does the document pack match the exact model and claims?» |
| Канада | Здоровье Канады Мдл where the product is a licensed medical device; device name, licence number, class, identifiers. | Электробезопасность, EMC, маркировка, importer and general product compliance review where applicable. | «Is there an active MDL for this exact device family or model? Which device identifiers are shown in MDALL?» |
| Австралия / NZ | TGA ARTG inclusion where applicable; sponsor, manufacturer, product type, class, ARTG ID. | RCM / EESS / ПОГОДА / Глобальный Марк evidence, электробезопасность, EMC, adapter / plug files, маркировка. | «Is this an ARTG medical-device entry or an electrical-safety / RCM document pack? Who is the sponsor or responsible supplier?» |
| Multi-market Private Label | Market-specific authorization or conformity route for each market; claim and labeling review by region. | CE / UKCA / FCC / RCM / ПОГОДА / Глобальный Марк / Rohs / EMC files by configuration, plus private-label artwork and manual review. | «Which documents can survive brand-name, упаковка, руководство, claim, accessory, adapter, or controller changes?» |
The practical rule: do not ask a supplier, “Do you have all certificates?” Ask, “Which files support this exact model, this claim, this accessory set, and this target market?»
RFQ Questions You Can Copy Before Contacting a Supplier
A serious supplier should answer with document names, certificate numbers, model scope, and clear boundaries, not only logo screenshots.
Use or adapt the questions below when comparing OEM / ODM suppliers:
| RFQ Question | What a Useful Answer Should Include |
|---|---|
| Is this exact model positioned as a medical / therapeutic device or a general beauty electrical appliance? | Использование по назначению, целевой рынок, claim boundary, и document route used for that positioning. |
| If FDA clearance is claimed, what is the 510(k) number and which models are covered? | K number, applicant, device name, product codes, decision date, и clearance letter / summary scope. |
| If the product is only FDA registered or listed, can you confirm it is not the same as FDA 510(k) прозрачный? | Establishment / listing context and careful compliant wording the brand should use. |
| Which documents cover EU sales: general CE documentation or CE marking under MDR? | EU Declaration of Conformity (DoC), applicable legislation / стандарты, and classification or MDR route if relevant. |
| Which Health Canada MDL or TGA ARTG records apply to this product? | Licence / ARTG ID, sponsor or manufacturer, product name, class, device family, and exact identifiers. |
| Do ISO 13485 and MDSAP certificates cover the manufacturing scope for this product category? | Certificate number, issuer, точный scope wording, manufacturing site, effective date, and expiry date. |
| Do electrical-safety or EMC files cover the whole device or only the adapter / charger? | Report or certificate number, standards applied, testing lab, covered model list, and verification of adapter, charger, батарея, контроллер, and accessory scope. |
| Are IEC 62471, Длина волны, and irradiance reports available for this exact model? | Report date, test distance, precise test conditions, sample ID, measured peak wavelength, и irradiance data. |
| If we change the brand, упаковка, руководство, claims, контроллер, charger, или аксессуары, which documents need review? | Private-label boundaries, artwork / IFU review steps, and document update/transfer requirements. |
| Which claims can we safely use, and which claims need regulatory or legal review? | Красота / wellness wording limits, medical/therapeutic wording boundaries, and the clinical/scientific evidence required for higher-risk claims. |
If a supplier cannot answer these questions, that does not automatically mean the product is bad. It does mean your procurement, согласие, or distributor team should slow down before paying for molds, упаковка, or a first production order.
Часто задаваемые вопросы
Is FDA registration the same as FDA 510(k) прозрачный?
Нет. FDA registration or device listing is not the same as FDA 510(k) прозрачный. А 510(k) clearance is tied to a specific device or device family and is based on substantial equivalence to a legally marketed predicate device.
Делает 510(k) clearance mean FDA approval?
Нет. FDA 510(k) clearance is not FDA approval. The safer wording is “FDA-cleared” or “510(k)-очищен,” not “FDA-approved,” unless the product actually has FDA approval through the appropriate pathway.
Can a private-label brand use a manufacturer’s 510(k)?
It depends on the device, маркировка, брендинг, distribution model, and regulatory responsibilities. A manufacturer-held clearance can be valuable, but private-label use should be reviewed with the manufacturer and regulatory counsel before launch.
Does one Health Canada MDL number cover every LED therapy device?
Нет. Buyers should review each MDALL licence record separately. For Sungrow, MDALL shows the manufacturer page under Company ID 187745, with separate records such as LED LIGHT THERAPY MASK and THE E-SERIES LED THERAPY LIGHT. Do not treat one MDL number as coverage for every product category.
Is TGA ARTG the same as FDA 510(k) or Health Canada MDL?
Нет. TGA ARTG, FDA 510(k), and Health Canada MDL are different market-specific regulatory records. A product may have documentation for one market and still need separate review or documentation for another market.
Do ISO 13485 and MDSAP mean the product itself is cleared?
Нет. Iso 13485 and MDSAP are quality-system evidence. They help buyers evaluate whether the manufacturer operates under a medical-device quality framework, but they do not replace product-level market authorization such as FDA 510(k), Health Canada MDL, or TGA ARTG.
Does UL 60601-1 или МЭК 60601 prove clinical effectiveness?
Нет. UL 60601-1 и МЭК 60601-1 relate to medical electrical safety and essential performance when those standards apply to the device. They do not prove treatment effectiveness and do not replace market authorization.
Are medical LED therapy devices and general LED beauty electrical devices certified the same way?
Нет. A medical or therapeutic LED device may need market authorization and medical-device quality evidence, depending on the target market and claims. A general beauty electrical appliance may mainly need CE / UKCA, FCC, Rohs, EMC, RCM / EESS, ПОГОДА / Глобальный Марк, adapter, electrical-safety, маркировка, and product-safety documentation. The claim and target market determine the document pack.
Is IEC 62471 the same as irradiance or wavelength testing?
Нет. МЭК 62471 is a photobiological safety evaluation for lamps and lamp systems. Wavelength and irradiance reports are separate optical-output records. For sourcing, buyers should request both safety reports and recent optical test data with measurement conditions.
What should I ask an LED therapy device OEM supplier for first?
Start with the target market and intended product category. Then request the 510(k) number if FDA clearance is claimed, Iso 13485 and MDSAP certificates, market-registration records such as Health Canada MDL or TGA ARTG where relevant, UL / МЭК 60601 electrical safety evidence where applicable, МЭК 62471 reports, and recent wavelength and irradiance test records.
What Should Buyers Do After Verifying the Certificates?
Once public records and certificate numbers are verified, the next step is to match the evidence to the product you actually plan to buy.
Before approving a supplier, confirm five things together:
- The exact model and product family.
- The controller, adapter, charger, батарея, аксессуары, and tested configuration.
- The target market and sales channel.
- The intended claims, маркировка, and private-label changes.
- The optical and production records available for the approved version.
Certification does not choose the right SKU, and a valid document does not automatically cover every configuration or marketing claim.
If you are still comparing mask platforms, use the LED Face Mask OEM Sourcing Guide. If the product model is already selected, continue with the relevant market, quality-system, electrical-safety, optical-safety, or claims guide in the Certification Topic Hub below.
Краткое содержание
LED therapy device certification is not one document. A serious OEM review should separate market authorization, quality-system certification, электробезопасность, фотобиологическая безопасность, optical output data, and factory background.
For buyers, the practical rule is simple: ask what each certificate proves, what it does not prove, and where it can be verified. A supplier with strong evidence should be able to provide public database links, certificate numbers, протоколы испытаний, model scope, and claim-review support without turning every document into a marketing claim.
If you are comparing Wakelife as a potential OEM partner, start with the relevant product category, then review the supporting documentation path below.
Certification Topic Hub
Use these deeper guides when you need to check one evidence layer in more detail:
| Тема | What it helps verify |
|---|---|
| FDA Registered vs FDA 510(k) Cleared LED Face Masks | The difference between FDA establishment registration, device listing, и 510(k) прозрачный |
| Health Canada MDL Verification for LED Therapy Devices | How to read MDALL records by manufacturer, licence number, class, device name, and identifier |
| TGA ARTG Verification for LED Face Masks and Red Light Therapy Devices | How to check ARTG ID, sponsor, manufacturer, product name, and device class |
| Iso 13485 vs MDSAP for LED Therapy Device Manufacturers | What quality-system certificates can prove, and what they cannot prove |
| МЭК 62471, Излучение, and Wavelength Reports for LED Face Masks | How to separate photobiological safety evidence from optical-output data |
| UL 60601-1 и МЭК 60601 for LED Therapy Devices | How to review electrical-safety evidence, file numbers, tested models, and covered accessories |
| CE, UKCA, FCC, SAA and Global-Mark Compliance for LED Therapy Devices | How to separate medical-device evidence from general beauty electrical, EMC, and regional market-access documents |
| CE vs CE MDR for LED Beauty Devices | How to distinguish general CE conformity for beauty electrical devices from CE marking under EU MDR for medical LED devices |
| LED Beauty Device Claims Guide | How beauty, Жестирочное, therapeutic, and medical wording changes the evidence and certification path |
Related Wakelife Resources
| Resource | Best use |
|---|---|
| OEM / ODM Services | Understand how documentation, настройка, and product development fit into an OEM project |
| Исследовать & Разработка | Review how engineering, тестирование, and compliance planning enter product development |
| Частная марка | Compare private-label sourcing with deeper OEM / ODM-разработка |
| Производство | Review factory background, production capability, and in-house manufacturing context |
| Светодиодная маска для лица | Explore the mask category most closely tied to FDA 510(k) K250830 and MDL 113279 |
| Панель светодиодной лампольной терапии | Explore panel and therapy-light categories related to broader MDL, TGA, and electrical-safety review |
| Лазерный светодиодный рост волос | Review a higher-compliance adjacent wearable category |
| Терапия красным светом для домашних животных | Review another light-therapy category where claims and documentation need careful separation |